Transcription factor binding sites in Baker’s yeast
M Hallett
21/07/2020
Where we are
Transcription factors bind DNA
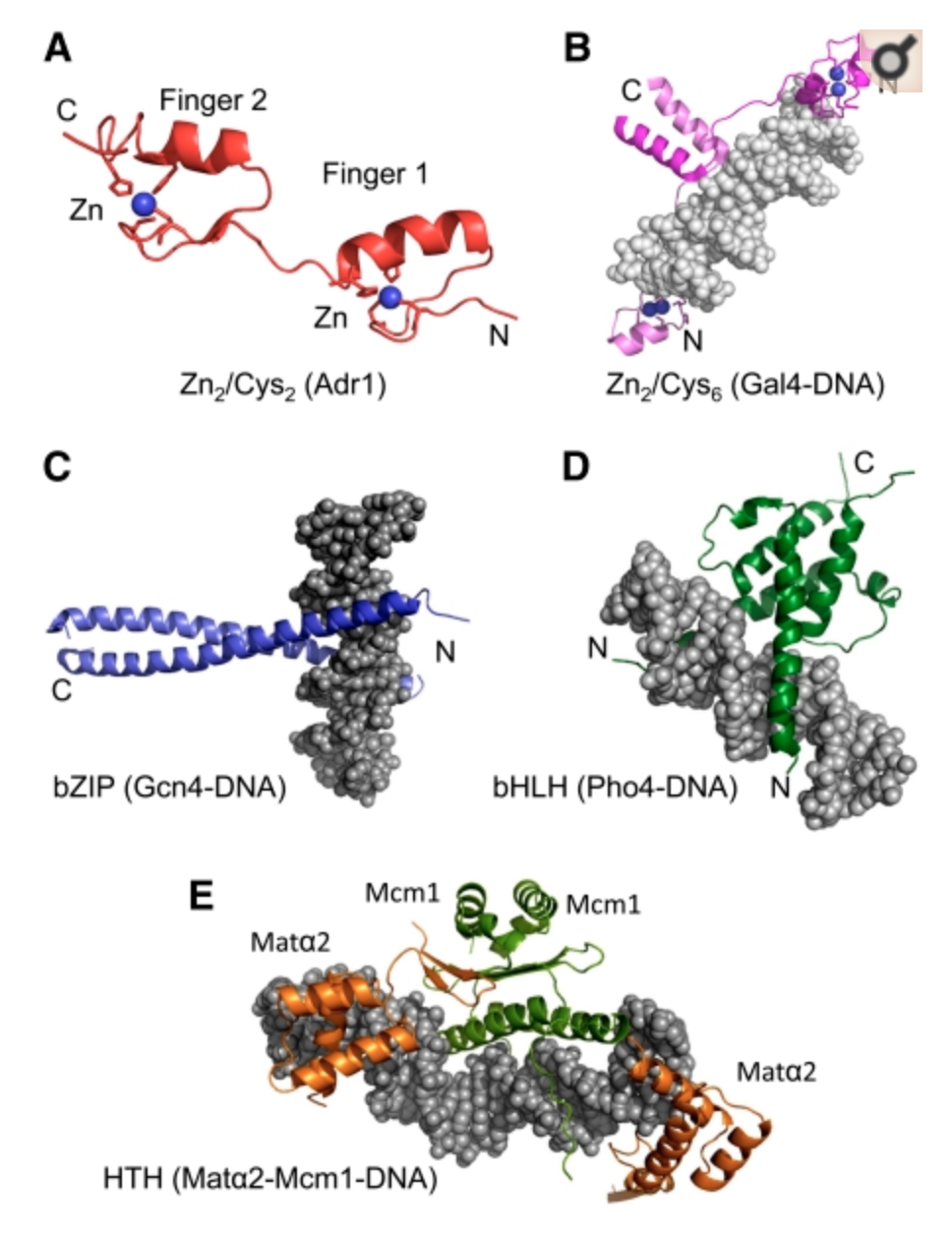
Distinct families of DBDs in Yeast
- In yeast, (most) TFs bind DNA using degenerate versions of these five families of protein stuctures.
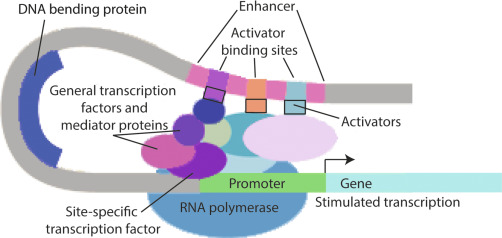
Transcription factor binding sites
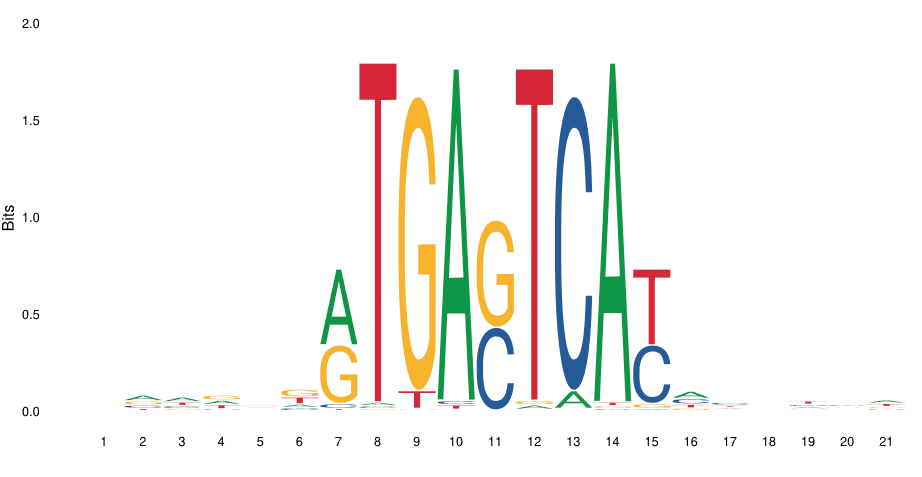
The DBD binds DNA in a semi-specific manner. In other words, the location where the TF binds DNA via the DBD is characterized by a degenerate motif. There is no one single nucleic acid sequence necesssary and sufficieint for DNA binding. Rather there is a “family” of related nucleic acid sequences, a degenerate motif.
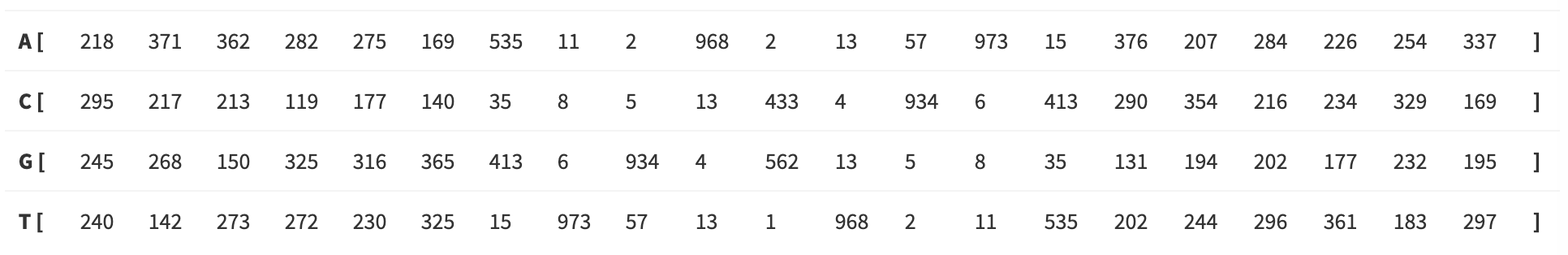
This is the binding motif for GCN4, a basic leucine ziper factor (bZIP) in yeast. You can see that some positions are highly determined (indices 7, 10, 12, 14) while others are variable (indices 7, 9, 11, 12, 15).
BIOCHEM xxxx
© M Hallett, 2022 Western University