02-solution
0001/01/01
\({\bf 67}\) total marks.
Question 1 [10 points] A. Re-express the following small program using pipes.
c <- 18
f <- c*(9/5) + 32
print(f)
## [1] 64.4
f <- c %>% .[[1]]*(9/5) + 32
print(f)
## [1] 64.4It’s pretty trick but just remember that whatever you pass through a pipe is called \({\tt .}\) and it’s actually a list. So if it is just a single item, it’s the first item in the list \({\tt .[[1]]}\)
B.
x <- 1e10
y <- sqrt( factorial(x) )
y <- x %>% factorial %>% sqrt %>% print
## [1] InfC.
x<-3; mu<-5; sigma <-7
nrm <- ( 1 / (sigma * sqrt( 2 * pi ))) * exp( -(1/2) * ( (x - mu)/ sigma )^2)
p <- c(x = 3, mu = 5, sigma = 7)
p %>%
{ ( 1 / (.[["sigma"]] * sqrt( 2 * pi ))) * exp( -(1/2) * ( (.[["x"]] - .[["mu"]])/ .[["sigma"]] )^2 ) }
## [1] 0.05471239So the trick is to combine the multiple parameters into a single vector first! That single vector is passed down the epipe.
p <- c( first = "a", second = "b", third = "c")D. What is this equation?
Normal probablity distribution function.
E.
site <- 15
gsub(";", "/", paste(paste( paste( "~hallett", paste("raw", "tara", sep = ";"), sep = ";"),
paste("site", site, sep="_"), sep=";"),
"dna_seq.fa", sep=";"))
## [1] "~hallett/raw/tara/site_15/dna_seq.fa"
site %>%
paste("site", ., sep="_") %>%
paste("dna_seq.fa", sep=";") %>%
paste("raw", "tara", ., sep = ";") %>%
paste("~hallett", ., sep = ";") %>%
gsub(";", "/", .)
## [1] "~hallett/raw/tara/site_15/dna_seq.fa"The pipes makes this one a bit easier to read; do you agree?
Question 2 [5 points]
In total, how many black, african or asian women are included in the \({\tt small\_brca}\) dataset?
q2 <- small_brca %>% filter(gender=="FEMALE", race=="BLACK OR AFRICAN AMERICAN" | race=="ASIAN") %>%
group_by(participant) %>% distinct %>% nrow
print(q2)## [1] 249Create a histogram of all asian individuals grouped by age. Each bar in the histogram should have two colours corresponding to male and females. (For example, red might be used for the fraction of women in some age group while blue is used for men in that same age group). Choose the number of bins (or binsize) in a way that makes your plot informative.
small_brca %>%
filter(race == "ASIAN") %>%
distinct(participant, .keep_all = TRUE) %>%
mutate(age_at_diagnosis = as.numeric(age_at_diagnosis)) %>%
ggplot(aes(x = age_at_diagnosis, fill = gender)) +
geom_histogram(binwidth = 5) So there are no males that fit this criterion. That’s ok. Also, you could use \({\tt birth\_days\_to}\) but you should divide by 365 first and take the absolute value (to make it more readable). Which to use is a bit of an esoteric issue: are you interested in their age at diagnosis or at last followup appointment?
So there are no males that fit this criterion. That’s ok. Also, you could use \({\tt birth\_days\_to}\) but you should divide by 365 first and take the absolute value (to make it more readable). Which to use is a bit of an esoteric issue: are you interested in their age at diagnosis or at last followup appointment?
Question 3 [7 points]
How many participants are there? (Careful: not samples but participants.) Show code.
small_brca %>% .["participant"] %>% distinct %>% nrow## [1] 1092A non-tidy way to do this would be as follows:
unique(small_brca$participant) %>% length## [1] 1092Both are perfectly ok. Do you undestand why we use \({\tt nrow}\) above but \({\tt length}\) in the non-tidy version?
List all of the \({\tt participant}\) that have more than one sample. Show code.
num_multi <- small_brca %>% group_by(participant) %>% summarise(count=n(), tumor, ESR1) %>%
filter(count > 1) %>% arrange(participant) ## `summarise()` has grouped output by 'participant'. You can override using the `.groups` argument.num_multi %>% nrow %>% print## [1] 238For the participants identified in 4b, create a scatter plot with the tumor on the \(x\) axis and the normal on the \(y\) axis (as specified by the \({\tt tumor}\) variable in the tibble) with points representing the expression of gene \({\tt ESR1}\).
for_plot <- num_multi %>% select(-count) %>%
pivot_wider(names_from = tumor, values_from = ESR1, values_fn=mean) %>% rename(tumor='TRUE', normal='FALSE')
ggplot(data = for_plot, mapping = aes(x = tumor, y = normal)) +
geom_point()## Warning: Removed 3 rows containing missing values (geom_point).
So that is some tricky coding! If you managed that, congratulations. Do you understand why the \({\tt values\_fn}\) is necessary? Here I have chosen the \({\tt mean}\) (average) to combine when a patient has more than one normal or tumor samples. In such a dataset, can you suggest why it might be advantageous to have more than one sample for tumor or normal for a patient? I included the \({\tt arrange}\) and \({\tt rename}\) calls just to make things a bit more readable; they aren’t strictly necessary.
Question 4 [23 points]
Show your code for each step below.
When I made the assignment, I had intended to put this question as Question 2. I was a bit sloppy. It should be easy by now!
Part a [1 point] Load the \({\tt tidyverse}\) library and load the \({\tt small\_brca}\) dataset.
Part b [1 point] Write code that determines the number of rows (observations) and columns (variables) in \({\tt small\_brca}\).
Part c [1 point] Write code that displays the column names (names of variable) for columns \(21\) to columns \(24\) inclusive.
Part d [2 points] What is the mean expression level of FOXC1?
Part e [3 points] How many samples are there with FOXC1 expression levels above \(245\)?
Part f [5 points] Create a new tibble called \({\tt bw}\) that contains only those rows (observations) from \({\tt small\_brca}\) that correspond to tumor samples of BLACK OR AFRICAN AMERICAN women.
Part g [5 points] Using \({\tt small\_brca}\), select the variables \({\tt participant}\), \({\tt vital\_status}\), and all genes except \({\tt RRM2}\).
Part h [5 points] Create a new variable called \({\tt triplenegative}\) in the tibble \({\tt small\_brca}\) that is TRUE (logical) if the sample is “Positive” for \({\tt er\_status\_by\_ihc}\), and not “Positive” for \({\tt her2\_fish\_status}\). Otherwise, in all other cases it is FALSE.
#a
library(tidyverse) # dont' need to install it; it's already been installed by MH
load("~/data/GDC/TCGA_BRCA/small_brca.Rdata")
#b
(dim(small_brca)) # one way to do it.## [1] 1215 79(nrow(small_brca))## [1] 1215(ncol(small_brca))## [1] 79#c
colnames(small_brca)[21:24]## [1] "er_status_by_ihc" "er_status_ihc_Percent_Positive"
## [3] "pr_status_by_ihc" "pr_status_ihc_percent_positive"#d
small_brca %>% summarize( my_mean= mean(FOXC1, na.rm=TRUE) ) %>% .[['my_mean']] %>% print## [1] 1717.955#e
small_brca %>% filter( FOXC1 > 245 ) %>% nrow## [1] 909#f
bw <- small_brca %>% filter( race=="BLACK OR AFRICAN AMERICAN", gender=="FEMALE", tumor==TRUE )
#g
small_brca %>% select( participant, vital_status, ANLN:TMEM45B ) %>% select( -RRM2 )## # A tibble: 1,215 x 51
## participant vital_status ANLN FOXC1 CDH3 FGFR4 UBE2T NDC80 PGR BIRC5
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 A1NF Alive 2124 235 485 261 1002 1184 710 1242
## 2 A27M Alive 4679 2068 15686 150 634 856 99 3429
## 3 A0GZ Alive 1192 284 3999 171 1314 732 8793 1738
## 4 A18V Dead 10191 2981 27406 473 1720 1853 76 4542
## 5 A13G Alive 823 142 435 46 77 45 8804 152
## 6 A275 Alive 10198 265 6903 8835 2997 1887 76 5517
## 7 A0XS Alive 669 294 1407 159 305 352 57567 673
## 8 A5RW Alive 1342 5638 2975 195 2275 2572 12 5022
## 9 A5RX Alive 420 531 5030 327 509 122 19520 284
## 10 A1RD Alive 1124 189 299 649 679 550 21586 982
## # … with 1,205 more rows, and 41 more variables: ORC6 <dbl>, ESR1 <dbl>,
## # PHGDH <dbl>, PTTG1 <dbl>, MELK <dbl>, NAT1 <dbl>, CXXC5 <dbl>, BCL2 <dbl>,
## # GPR160 <dbl>, EXO1 <dbl>, UBE2C <dbl>, TYMS <dbl>, KRT5 <dbl>, KRT14 <dbl>,
## # MAPT <dbl>, CDC6 <dbl>, MMP11 <dbl>, MYBL2 <dbl>, SFRP1 <dbl>, CCNE1 <dbl>,
## # BLVRA <dbl>, BAG1 <dbl>, MLPH <dbl>, CDC20 <dbl>, CENPF <dbl>, MIA <dbl>,
## # KRT17 <dbl>, FOXA1 <dbl>, ACTR3B <dbl>, CCNB1 <dbl>, MDM2 <dbl>, MYC <dbl>,
## # CEP55 <dbl>, SLC39A6 <dbl>, ERBB2 <dbl>, GRB7 <dbl>, KIF2C <dbl>,
## # NUF2 <dbl>, EGFR <dbl>, MKI67 <dbl>, TMEM45B <dbl>#h within the tidyverse
small_brca <- small_brca %>% mutate( triplenegative = ifelse(er_status_by_ihc=="Positive", TRUE , FALSE ) )
#h - non-tidy verse way
small_brca$triplenegative <- ifelse(small_brca$er_status_by_ihc=="Positive", TRUE , FALSE )Note that when I work on my machine at home, the path is different. Your path for the \({\tt load}\) should look something like \(data/small_brca.Rdata\) because you are on RStudio Cloud and your directory structure looks the same. That’s one of the advantages of RStudio Cloud: you don’t need to struggle with paths on your own machine as much.
There are different ways to do part (d). For example, in a none-tidyverse way:
( mean( as.numeric(small_brca$FOXC1 ), na.rm=TRUE ) )## [1] 1717.955In both cases, do you understand the reason for \({\tt na.rm}\)? Also, the \({\tt as.numeric}\) is necessary because when you use the \({\tt \$}\) to select a column, it is still a tibble (it’s just a single column tibble). You need to turn that tibble into a vector of numbers. Make sure you understand the \({\tt ifelse}\) function; it’s handy.
Question 5 [12 points]
Using the \({\tt small\_brca}\) tibble, create the following plot.
ggplot(data = small_brca) +
geom_point(mapping = aes(x = GRB7, y = EGFR, color=tumor)) +
facet_wrap(~ tumor_status, nrow = 2) +
scale_y_continuous(trans='log') + scale_x_continuous(trans='log') +
ggtitle("GRB7 and EGFR expression by tumor_status") +
xlab("log GRB7 expression") + ylab("log EGFR expression")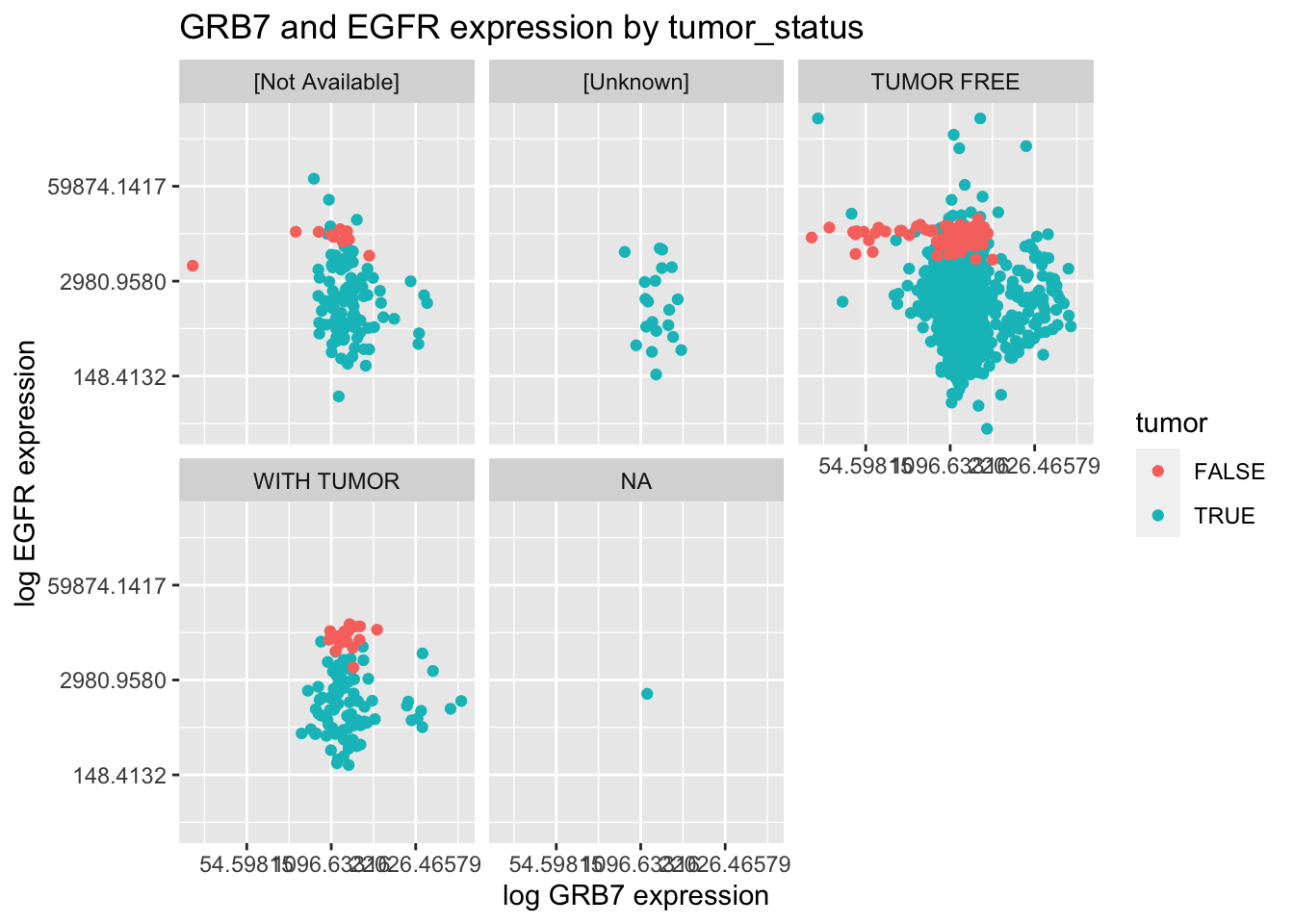
Question 6 [5 points]
The variable \({\tt race}\) in \({\tt small\_brca}\) has several different possible values including “WHITE”, “ASIAN”, “BLACK OR AFRICAN AMERICAN”, “AMERICAN INDIAN OR ALASKA NATIVE”, “[Not Available]”, “[Not Evaluated]”, or NA.
It is better to simply use NA instead of“[Not Available]”, “[Not Evaluated]”, or NA. It will simplify your code in downstream analyses.
Write R code to create a new \({\tt race\_modified}\) variable where all samples with “[Not Available]”, “[Not Evaluated]” are changed to NA (Note: you shouldn’t use “NA”, but NA. They are not the same)
print(unique(small_brca$race))## [1] "WHITE" "ASIAN"
## [3] "BLACK OR AFRICAN AMERICAN" "[Not Available]"
## [5] "[Not Evaluated]" NA
## [7] "AMERICAN INDIAN OR ALASKA NATIVE"small_brca <- small_brca %>% mutate(race_modified = ifelse((race=="[Not Available]" |
race=="[Not Evaluated]" |
race=="NA"), NA, race))
print(unique(small_brca$race_modified))## [1] "WHITE" "ASIAN"
## [3] "BLACK OR AFRICAN AMERICAN" NA
## [5] "AMERICAN INDIAN OR ALASKA NATIVE"Question 7 [5 points] Pick one of the \(50\) genes from \({\tt small\_brca}\) uniformly randomly (see the \({\tt runif}\) function). Using the NCBI, provide the following information or note that it is not availalbe:
colnames(small_brca)[runif(1, min=37, max=79)] %>% print## [1] "TYMS"The rest of the answers for Question 7 are specific to the invividual.
10a Full name of the gene and the official name according to the HGNC.
10b First time it was reported in the literature.
10c Where it is located in the genome.
10d Its \({\tt gi}\) acccession code or codes (if it has been modified).
10e The number of exons.
10f The number of alternative transcripts that have been record.
10g Its full amino acid sequence. Provide its protein ID.
10h Its protein structure, if known.